
I. Przedmiot opinii
Przedmiotem niniejszej opinii, sporządzonej przez prawników ze Stowarzyszenia Prawników Głos Wolności z siedzibą w Katowicach, jest udzielenie odpowiedzi na pytanie, czy szczepienia przeciwko COVID-19 stosowane w ramach Narodowego Programu Szczepień są eksperymentem medycznym.
W opinii przedstawiono kolejno: podstawę prawną i faktyczną rozważań (pkt II i III niżej), analizę (pkt IV niżej) oraz wnioski (pkt V).
II. Podstawa prawna
- Konstytucja Rzeczypospolitej Polskiej z dnia 2 kwietnia 1997 r. (Dz. U. Nr 78, poz. 483 z późn. zm.), zwana dalej “Konstytucją RP”;
- ustawa z dnia 5 grudnia 1996 roku o zawodach lekarza i lekarza dentysty (Dz. U. 2021.790, t.j. z dnia 28.04.2021 r.), zwana dalej “U.z.l.”;
- ustawa z dnia 6 września 2001 roku – Prawo farmaceutyczne (Dz. U. 2021.974, t.j. z dnia 28.05.2021 r.), zwana dalej “Prawem farmaceutycznym”
- ROZPORZĄDZENIE KOMISJI (WE) NR 507/2006 z dnia 29 marca 2006 r. w sprawie warunkowego dopuszczenia do obrotu produktów leczniczych stosowanych u ludzi wchodzących w zakres rozporządzenia (WE) nr 726/2004 Parlamentu Europejskiego i Rady, zwane dalej „Rozporządzeniem KOMISJI (WE) NR 507/2006 z dnia 29 marca 2006 r.”
III. Stan faktyczny
W przestrzeni publicznej, w tym na stronach rządowych oraz w oficjalnych komunikatach podawanych przez media wskazuje się, że szczepienia przeciw COVID-19 prowadzone w ramach Narodowego Programu Szczepień nie stanowią eksperymentu medycznego w rozumieniu U.zl. oraz Prawa farmaceutycznego. Tym samym, powstała konieczność zweryfikowania, czy rzeczywiście ww. komunikaty medialne znajdują potwierdzenie w obowiązujących przepisach prawa.
IV. Analiza merytoryczna
- Pojęcie eksperymentu medycznego w U.z.l. oraz w Konstytucji RP
W pierwszej kolejności należy wskazać, w jaki sposób prezentuje się w sensie jurydycznym pojęcie eksperymentu medycznego.
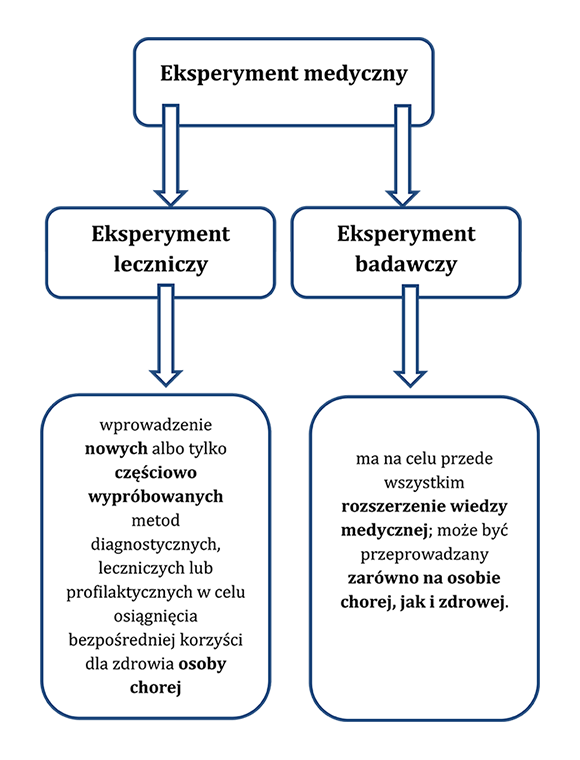
Zgodnie z art. 21 ust. 1 U.z.l. eksperyment medyczny przeprowadzany na ludziach może być eksperymentem leczniczym albo eksperymentem badawczym.
Zgodnie z art. 21 ust.2 U.z.l. eksperymentem leczniczym jest wprowadzenie nowych albo tylko częściowo wypróbowanych metod diagnostycznych, leczniczych lub profilaktycznych w celu osiągnięcia bezpośredniej korzyści dla zdrowia osoby chorej. Może on być przeprowadzony, jeżeli dotychczas stosowane metody nie są skuteczne albo jeżeli ich skuteczność nie jest wystarczająca. Udział w eksperymencie leczniczym kobiet ciężarnych wymaga szczególnie wnikliwej oceny związanego z tym ryzyka dla matki i dziecka poczętego.
Zgodnie z art. 21 ust. 3 U.z.l. eksperyment badawczy ma na celu przede wszystkim rozszerzenie wiedzy medycznej. Może być on przeprowadzany zarówno na osobie chorej, jak i zdrowej. Przeprowadzenie eksperymentu badawczego jest dopuszczalne, gdy uczestnictwo w nim nie jest związane z ryzykiem albo też ryzyko jest minimalne i nie pozostaje w dysproporcji do możliwych pozytywnych rezultatów takiego eksperymentu.
Ponadto, zgodnie z art. 22 U.z.l. eksperyment medyczny może być przeprowadzany, jeżeli spodziewana korzyść lecznicza lub poznawcza ma istotne znaczenie, a przewidywane osiągnięcie tej korzyści oraz celowość i sposób przeprowadzania eksperymentu są zasadne w świetle aktualnego stanu wiedzy i zgodne z zasadami etyki zawodu medycznego.
Zgodnie z art. 39 Konstytucji RP nikt nie może być poddany eksperymentom naukowym, w tym medycznym, bez dobrowolnie wyrażonej zgody. Co więcej, zgodnie z art. 23 a) ust. 1 pkt 3 U.z.l. zabrania się przeprowadzania eksperymentu badawczego na żołnierzu i innej osobie pozostającej w zależności hierarchicznej ograniczającej swobodę dobrowolnego wyrażania zgody.
Wskazane w tym przepisie pojęcie “eksperymentu medycznego” jest w powszechnie obowiązującej wykładni rozumiane jako rodzaj eksperymentu naukowego najsilniej ingerującego w integralność psychofizyczną człowieka, który służy bezpośrednio ratowaniu życia ludzkiego, a jego potencjalne korzyści mają dotknąć osobę poddaną temu eksperymentowi. Może on zostać przeprowadzony, jeżeli dotychczasowe metody lecznicze są nieskuteczne lub skuteczne w stopniu niewystarczającym. (Tuleja Piotr (red.), Konstytucja Rzeczypospolitej Polskiej. Komentarz, wyd. II).
Na uwagę zasługuje także pojęcie pojawiające się w art. 2 pkt 2 Prawa farmaceutycznego, mówiące o badaniu klinicznym, gdzie z definicji tego pojęcia wynika jednoznacznie, że jest ono przeprowadzane z udziałem ludzi i jego celem jest zbadanie lub potwierdzenie działalności skutków farmakologicznych produktu leczniczego, zaś sam produkt leczniczy obejmuje swoim zakresem również wszelkiego rodzaju szczepionki, co wynika z treści definicji produktu leczniczego, zgodnie z art. 2 pkt 30 w zw. z art. 2 pkt 32 Prawa farmaceutycznego.
Zgodnie z art. 39 Konstytucji RP nikt nie może być poddany eksperymentom naukowym, w tym medycznym, bez dobrowolnie wyrażonej zgody.
Wskazane w tym przepisie pojęcie “eksperymentu medycznego” jest w powszechnie obowiązującej wykładni rozumiane jako rodzaj eksperymentu naukowego najsilniej ingerującego w integralność psychofizyczną człowieka, który służy bezpośrednio ratowaniu życia ludzkiego, a jego potencjalne korzyści mają dotknąć osobę poddaną temu eksperymentowi. Może on zostać przeprowadzony, jeżeli dotychczasowe metody lecznicze są nieskuteczne lub skuteczne w stopniu niewystarczającym. (Tuleja Piotr (red.), Konstytucja Rzeczypospolitej Polskiej. Komentarz, wyd. II).
Konstytucja RP, choć chroni wolność prowadzenia badań naukowych (art. 73 Konstytucji RP), nie traktuje jej jako wolności absolutnej, a jej granice wyznacza między innymi obowiązek poszanowania godności człowieka (art. 30 Konstytucji RP). Godność ta w różnych zresztą aspektach mogłaby zostać naruszona przez eksperymenty naukowe i medyczne sprowadzające człowieka jedynie do roli przedmiotu tych badań.
- Brak zakończonych badań klinicznych nad szczepionkami przeciwko COVID-19
Ocenę, czy szczepienia przeciwko COVID-19 są eksperymentem medycznym, czy też nie, należy rozpocząć od analizy art. 2 pkt 2 oraz art. 37a ust. 2 Prawa farmaceutycznego, zgodnie z którymi:
| Art. 2 pkt 2 Prawa farmaceutycznego: Badaniem klinicznym jest każde badanie prowadzone z udziałem ludzi w celu odkrycia lub potwierdzenia klinicznych, farmakologicznych, w tym farmakodynamicznych skutków działania jednego lub wielu badanych produktów leczniczych, lub w celu zidentyfikowania działań niepożądanych jednego lub większej liczby badanych produktów leczniczych, lub śledzenia wchłaniania, dystrybucji, metabolizmu i wydalania jednego lub większej liczby badanych produktów leczniczych, mając na względzie ich bezpieczeństwo i skuteczność. |
| Art. 37 a ust. 2 Prawa farmaceutycznego Badanie kliniczne produktu leczniczego jest eksperymentem medycznym z użyciem produktu leczniczego przeprowadzanym na ludziach w rozumieniu przepisów ustawy z dnia 5 grudnia 1996 r. o zawodach lekarza i lekarza dentysty (Dz. U. z 2021 r. poz. 790 i 1559), zwanej dalej „ustawą o zawodzie lekarza”. |
Tym samym, ocena prawna Narodowego Programu Szczepień musi uwzględniać, czy badania kliniczne nad szczepionkami przeciw COVID-19 zostały zakończone, czy też nie.
Należy zatem sięgnąć do Charakterystyki Produktu Leczniczego (CHPL) szczepionek przeciwko COVID-19.
- Według informacji wynikających z Charakterystyki Produktu Leczniczego (CHPL) firmy Pfizer znajdującej się na str. 15 wynika, że:
„W celu potwierdzenia skuteczności i bezpieczeństwa stosowania produktu leczniczego Comirnaty, podmiot odpowiedzialny powinien przedłożyć raport końcowy z badania klinicznego dla randomizowanego, z grupą kontrolną otrzymującą placebo, prowadzonego metodą ślepej próby wobec obserwatora badania C4591001. Termin: do grudnia 2023 r.”
- Według informacji wynikających z Charakterystyki Produktu Leczniczego (CHPL) firmy Moderna znajdującej się na str. 15 wynika, że:
„W celu potwierdzenia skuteczności i bezpieczeństwa stosowania COVID-19 Vaccine Moderna podmiot odpowiedzialny powinien przedłożyć raport końcowy z badania klinicznego dotyczący randomizowanego, kontrolowanego placebo, zaślepionego dla obserwatora badania klinicznego o numerze mRNA-1273-P301. Termin: do grudnia 2022 r.”
- Według informacji wynikających z Charakterystyki Produktu Leczniczego (CHPL) firmy Janssen znajdującej się na str. 20 wynika, że:
„W celu potwierdzenia skuteczności i bezpieczeństwa stosowania szczepionki Ad26.COV2.S przeciw COVID-19, podmiot odpowiedzialny powinien przedstawić końcowe sprawozdanie z randomizowanego, kontrolowanego placebo badania VAC31518COV3001 z zastosowaniem ślepej próby wobec obserwatora. Termin: 31 grudnia 2023 r.”
- Według informacji wynikających z Charakterystyki Produktu Leczniczego (CHPL) firmy AstraZeneca znajdującej się na str. 19 wynika, że:
„W celu potwierdzenia skuteczności i bezpieczeństwa stosowania szczepionki COVID-19 Vaccine AstraZeneca, podmiot odpowiedzialny powinien złożyć ostateczne raporty końcowe (ang. Clinical Study Reports, CSR) z randomizowanych, kontrolowanych badań klinicznych COV001, COV002, COV003 i COV005. Termin: 31 maja 2022 r.”
Z powyższych informacji bezsprzecznie wynika, że raporty końcowe dotyczące potwierdzenia skuteczności i bezpieczeństwa stosowania szczepionek przeciwko COVID-19 zostaną dopiero złożone. Tym samym, nie zakończono badań klinicznych nad ich stosowaniem.
- Warunkowe dopuszczenie do obrotu szczepionek przeciw COVID-19
Ponadto, wszystkie szczepionki przeciw COVID-19, wyprodukowane przez firmy BionTech-Pfizer, Moderna, Astrazeneca oraz Johnson and Johnson (Janssen) zostały objęte warunkowym pozwoleniem na dopuszczenie do obrotu, co oznacza, że są one stosowane jako szczepionki, których skutki farmakologiczne nie są do końca znane.
Pozwolenie na warunkowe dopuszczenie do obrotu wydawane jest w oparciu o ROZPORZĄDZENIE KOMISJI (WE) NR 507/2006 z dnia 29 marca 2006 r. w sprawie warunkowego dopuszczenia do obrotu produktów leczniczych stosowanych u ludzi wchodzących w zakres rozporządzenia (WE) nr 726/2004 Parlamentu Europejskiego i Rady, zgodnie z którym:
- W przypadku niektórych kategorii produktów leczniczych, aby spełnić niezaspokojone potrzeby lecznicze pacjentów oraz na rzecz zdrowia publicznego, może okazać się niezbędne przyznanie pozwoleń na dopuszczenie do obrotu na podstawie mniej kompletnych danych niż ma to miejsce zazwyczaj i przy zachowaniu szczególnych zobowiązań; takie pozwolenie jest dalej zwane „warunkowym pozwoleniem na dopuszczenie do obrotu” (pkt 2 Rozporządzenia KOMISJI (WE) NR 507/2006 z dnia 29 marca 2006 r.)
- Pomimo że dane, na których oparta jest opinia w sprawie warunkowego pozwolenia na dopuszczenie do obrotu, mogą być mniej kompletne, stosunek korzyści do ryzyka, jak określono w art. 1 pkt 28 lit. a) dyrektywy 2001/83/WE, powinien być dodatni. Ponadto korzyści dla zdrowia publicznego wynikające z natychmiastowej dostępności na rynku produktu leczniczego, którego to dotyczy, powinny przewyższać ryzyko związane z faktem, że wciąż wymagane są dodatkowe dane (pkt 3 Rozporządzenia KOMISJI (WE) NR 507/2006 z dnia 29 marca 2006 r.)
- Niezbędne jest, by pozwolenia na dopuszczenie do obrotu podlegały szczególnym zobowiązaniom. Podmiot odpowiedzialny powinien być zobowiązany do uzupełnienia lub podjęcia pewnych badań mających na celu potwierdzenie, że stosunek korzyści do ryzyka jest dodatni, a także do rozwiązania wszelkich kwestii dotyczących jakości, bezpieczeństwa i skuteczności produktu (pkt 5 Rozporządzenia KOMISJI (WE) NR 507/2006 z dnia 29 marca 2006 r.)
- Zgodnie z rozporządzeniem (WE) nr 726/2004 warunkowe pozwolenia na dopuszczenie do obrotu będą ważne przez rok z możliwością odnowienia (pkt 9 Rozporządzenia KOMISJI (WE) NR 507/2006 z dnia 29 marca 2006 r.)
- Pacjentom i specjalistom z zakresu opieki zdrowotnej należy dostarczyć jednoznaczne informacje dotyczące warunkowego charakteru pozwoleń. Niezbędne jest zatem, aby takie informacje były wyraźnie podane w charakterystyce produktu leczniczego, a także w ulotce informacyjnej dołączonej do produktu leczniczego (pkt 10 Rozporządzenia KOMISJI (WE) NR 507/2006 z dnia 29 marca 2006 r.)
Zgodnie z art. 4 Rozporządzenia KOMISJI (WE) NR 507/2006 z dnia 29 marca 2006 r.:
| Warunkowe pozwolenie na dopuszczenie do obrotu może zostać przyznane, jeżeli Komitet uzna, że mimo iż wyczerpujące dane kliniczne dotyczące bezpieczeństwa i skuteczności produktu leczniczego nie zostały dostarczone, spełnione są łącznie następujące wymagania:stosunek korzyści do ryzyka produktu leczniczego, jak określono w art. 1 pkt 28 lit. a) dyrektywy 2001/83/WE, jest dodatni;prawdopodobne jest, że wnioskodawca będzie w stanie dostarczyć wyczerpujące dane kliniczne;niezaspokojone potrzeby medyczne zostaną spełnione;korzyści dla zdrowia publicznego wynikające z natychmiastowej dostępności na rynku danego produktu leczniczego przewyższają ryzyko związane z faktem, że wymagane są dodatkowe dane. |
Warunkowy charakter dopuszczenia szczepionek do obrotu oraz wskazywane przez samą Komisję Europejską ryzyko powikłań rodzi wątpliwości natury prawnej w aspekcie sposobu przeprowadzania szczepień przeciwko COVID-19. W szczególności chodzi o dwie kwestie, tj.: o pełną wiedzę pacjentów w zakresie możliwości powstania negatywnych odczynów poszczepiennych (NOP) oraz o wyrażenie w powyższym aspekcie świadomej zgody pacjentów na przyjęcie szczepionki.
Zgoda na przeprowadzenie eksperymentu powinna być wyrażona zawsze indywidualnie, nawet gdyby eksperymenty medyczne dotyczyły łącznie pewnej grupy osób. Rzecz jasna, zgoda ta powinna nastąpić po udzieleniu informacji o szansach i zagrożeniach związanych z eksperymentem leczniczym. Racjonalne i zgodne z Konstytucją RP wydają się postanowienia U.z.l. przewidujące wymóg pisemnej zgody osoby badanej mającej w nim uczestniczyć, a w razie niemożności wyrażenia pisemnej zgody – wyrażenie zgody ustnie złożone w obecności dwóch świadków. Tak wyrażona zgoda powinna być odnotowana w dokumentacji lekarskiej.[1]
- Badanie kliniczne a badanie nieinterwencyjne
W literaturze przedmiotu pojawił się pogląd, że skoro zgodnie z art. 37 al pkt 4 Prawa farmaceutycznego przepisów rozdziału 2a Prawa farmaceutycznego pt. „Badania kliniczne produktów leczniczych” nie stosuje się do badań przeprowadzanych po wydaniu pozwolenia na dopuszczenie do obrotu oraz badań nieinterwencyjnych, to szczepienia przeciwko COVID-19 nie są eksperymentem medycznym.
Wynika to z błędnego założenia, że szczepionki przeciwko COVID-19 są tzw. badaniem nieinterwencyjnym, co powoduje zwolnienie ich od wymogów stawianych badaniu klinicznemu. Szczepionki przeciwko COVID-19 są powszechnie stosowane, pomimo że wciąż nie są znane ostatecznie skutki ich przyjęcia, jak również stosuje się je wyłącznie na osobach zdrowych. Co więcej doktryna wskazuje, że ze względu na szeroką definicję eksperymentu badawczego (będącego rodzajem eksperymentu medycznego) badania nieinterwencyjne, mimo że nie są badaniami klinicznymi, mogą wypełniać dyspozycję definicji eksperymentu badawczego zawartą w art. 21 ust. 3 U.z.l. W konsekwencji do takich badań znajdowałby zastosowanie art. 29 U.z.l., przewidujący wymóg uzyskania pozytywnej opinii niezależnej komisji bioetycznej. [2]
Kolejnym argumentem wskazującym, że Narodowy Program Szczepień spełnia przesłanki eksperymentu medycznego, jest ciągle istniejąca niepewność co skutków stosowania szczepionek przeciw COVID-19. Na przykładzie firmy Pfizer można wskazać, że wciąż brakuje informacji, jaki wpływ szczepionka ta ma na zdrowie kobiet w ciąży oraz także w jaki sposób wpływa (lub czy w ogóle) na mleko matki. Ponadto nie sposób określić, jakie skutki zostaną wywołane szczepieniem w stosunku do osób o obniżonej odporności.
- Kwestionariusz wypełniany przed podaniem szczepionki przeciwko COVID-19
Zgodnie z art. 37 b ust. 2 pkt 2 Prawa farmaceutycznego badanie kliniczne przeprowadza się, uwzględniając, że:
- prawa
- bezpieczeństwo
- zdrowie i dobro uczestników badania klinicznego
są nadrzędne w stosunku do interesu nauki oraz społeczeństwa, jeżeli w szczególności:
- uczestnik badania klinicznego, a w przypadku gdy osoba ta nie jest zdolna do wyrażenia świadomej zgody – jej przedstawiciel ustawowy, podczas przeprowadzonej przed badaniem klinicznym rozmowy z badaczem lub z członkiem jego zespołu, zapoznali się z celami, ryzykiem i niedogodnościami związanymi z tym badaniem klinicznym oraz warunkami, w jakich ma ono zostać przeprowadzone, a także zostali poinformowani o przysługującym ich prawie do wycofania się z badania klinicznego w każdej chwili;
Dokonując wykładni art. 37b ust. 2 pkt 2 Prawa farmaceutycznego, należy zwrócić uwagę na to, jaka jest relacja tego przepisu w stosunku do kwestionariusza, który każda osoba poddana szczepieniu przeciw COVID-19 musi wypełnić i opatrzyć swoim własnoręcznym podpisem. Kwestionariusz ten mówi wyłącznie o ocenie ryzyka wystąpienia powikłań poszczepiennych, jednakże nie jest to wystarczające, aby można było mówić o spełnieniu przesłanek, o których mowa w powyższym przepisie.
Składają się na nie:
- świadoma zgoda pacjenta (uczestnika badania klinicznego), która bezpośrednio wiąże się z uświadomieniem o celach, ryzyku i niedogodnościach związanych z danym badaniem oraz o warunkach w jakich zostanie przeprowadzone
- przestrzeganie prawa uczestnika badania klinicznego do zapewnienia jego integralności fizycznej i psychicznej, prywatności oraz ochrony danych osobowych
- możliwość wycofania się uczestnika z badania klinicznego
- uprzednie zawarcie pomiędzy sponsorem i badaczem umowę obowiązkowego ubezpieczenia odpowiedzialności cywilnej za szkody wyrządzone w związku z prowadzeniem badania klinicznego.
Żaden kwestionariusz wypełniany przed podaniem szczepionki przeciwko COVID-19 takich informacji nie zawiera.
V. Wnioski
Porównując zasady dotyczące przeprowadzania eksperymentu medycznego, wynikające z Prawa farmaceutycznego oraz U.z.l. z Narodowym Programem Szczepień, zasadne jest uznanie, że w przypadku tego programu mamy do czynienia z eksperymentem medycznym.
Wynika to z faktu, że badania nad określeniem skutków działalności szczepionek przeciw COVID-19 wciąż nie zostały zakończone. Ponadto, odpowiednie instytucje międzynarodowe jak Europejska Agencja Leków zastosowały uproszczoną procedurę warunkowego dopuszczenia do obrotu szczepień przeciwko COVID-19, pomimo braku zakończenia badań klinicznych nad tymi produktami. Uczyniono to po to, by umożliwić zastosowanie tych produktów na szeroką skalę.
Z definicji eksperymentu medycznego wskazanej w art. 39 Konstytucji RP, w art. 21 U.z.l. oraz w art. 2 pkt 2 w zw. z art. 37a ust.2 Prawa farmaceutycznego jednoznacznie wynika, że przeprowadzenie masowej akcji szczepień za pomocą szczepionek przeciw COVID-19 wypełnia definicję eksperymentu medycznego (w tym badawczego, ponieważ jego cele są zbieżne z celami, które stawiają sobie instytucje prowadzące masową akcję szczepień przeciw COVID-19).
Niewątpliwie, wszystkie dostępne w chwili obecnej szczepionki przeciwko COVID-19 ingerują w integralność zarówno psychiczną jak i fizyczną człowieka, gdyż powodują bezpośrednią styczność organizmu z dawką szczepionki i bezpośrednio na niego wpływają. Nie są przy tym znane skutki tych szczepionek dla szerokiego grona osób. Nie oceniano skuteczności, bezpieczeństwa stosowania ani immunogenności szczepionek u osób z obniżoną odpornością, w tym u pacjentów otrzymujących leczenie immunosupresyjne. Istnieje tylko ograniczone doświadczenie dotyczące stosowania szczepionek u kobiet w okresie ciąży, nie wiadomo, czy szczepionki przenikają do mleka ludzkiego. Nie przeprowadzono również żadnych badań nad genotoksycznością ani rakotwórczością szczepionek przeciwko COVID-19.